GLDP000155
In-House Research
Binding free energy analysis of galectin‐3 natural ligands and synthetic inhibitors
Chronicle Description
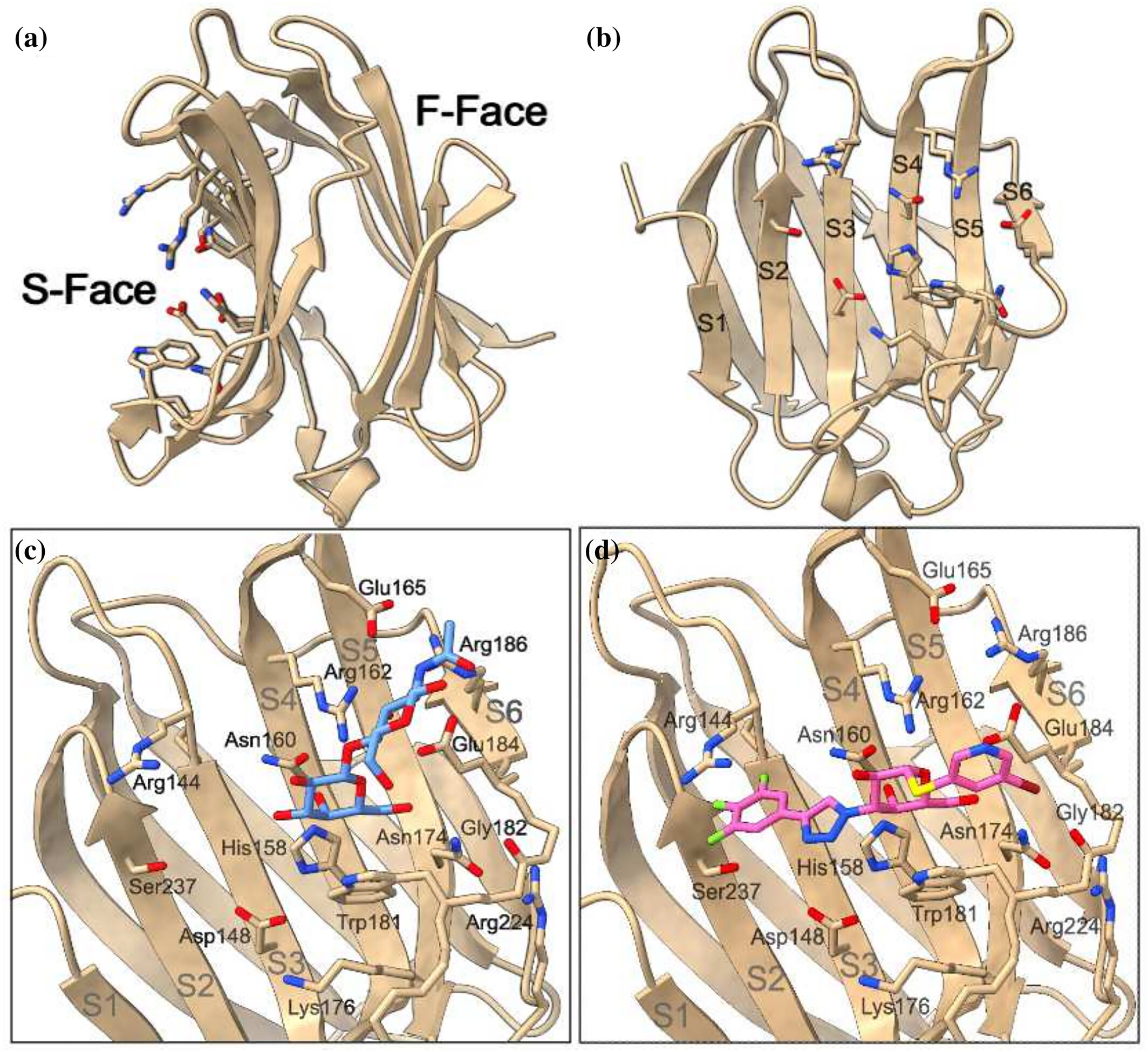
Galectin-3 -ligand complexes are characterized by halogen, σ -hole bonds, hydrogen bonds, cationπ and CHπ interactions. Here, we model these non-covalent interactions with the AMOEBA polarizable force field and conduct an absolute binding free energy analysis on leading galectin-3 inhibitors. Synthetic drug molecules GB0139, GB1107, and GB1211 were estimated to have binding free energies of /C0 4.3, /C0 6.7, and /C0 9.5 kcal/mol respectively. This compares to /C0 0.3 and 1.4 kcal/mol for the natural ligands, N-acetyllactosamine type 1 and type 2, respectively. We calculated the electric fields projected along key bonds in each ligand to further rationalize these results. We find that while the hydroxyl groups of the natural ligands interact reasonably well with residues in galectin-3 ' s binding pocket, structural dynamics weaken the binding pose and favor interactions with water, sometimes yielding to dissociation. In contrast, the more favorable binding energy of GB1211, leading inhibitor in clinical studies, is associated with strong and constant electric fields across the bonds investigated, suggesting a stiffer binding pose with a stabilizing σ -hole interaction.
Associated Publications
Luke Newman; Valerie Vaissier Welborn
Protein Science
Data Files & Attachments
1 File(s) Available
| File Name | Description | File Type | Size | Action |
|---|---|---|---|---|
| Supporting Information | Supporting Information | 3.96 MB | Login to Download |
Research Keywords & Tags
Related Chronicles